编辑说:近几年,国家层面支持中医药发展并未停歇,中药新药申报及获批数量屡创新高,激发起了国内中药研发企业寻求新突破,加快新药面世的步伐。6月27日在2024米思会——米交汇分会上,由盈科瑞创新医药协办的“中医药创新生态合作论坛”邀请到了多位重磅嘉宾,共同探讨“加强产业链融合,助力中医药创新成果转化”的话题。
精彩内容
近几年,国家层面支持中医药发展并未停歇,中药新药申报及获批数量屡创新高,激发起了国内中药研发企业寻求新突破,加快新药面世的步伐。6月27日在2024米思会——米交汇分会上,由盈科瑞创新医药协办的“中医药创新生态合作论坛”邀请到了多位重磅嘉宾,共同探讨“加强产业链融合,助力中医药创新成果转化”的话题。
本会议由中国中医科学院中药研究所资深研究员、北京盈科瑞创新医药股份有限公司董事长张保献主持,出席本次会议的专家学者有中国医学科学院药用植物研究所原所长孙晓波、香港浸会大学中医药研发中心科学顾问窦金辉、粤港澳大湾区药品与医疗器械真实世界研究院院长蒋杰、香港浸会大学药学博士&华药君康高级顾问黄韬以及北京盈科瑞创新医药股份有限公司科研总裁李艳英。
4000亿市场热火朝天,临床价值已成“最强核心”
米内网数据显示,在中国三大终端六大市场(统计范围见文末)中成药的合计销售规模连续三年保持正增长态势,2023年已突破4100亿元,增长率为6.51%,而西药(化药+生物药)的合计销售规模则呈现跌宕起伏的态势,2022年下滑了0.47%,2023年增长了4.67%,中成药市场潜力正在持续爆发,势不可挡。
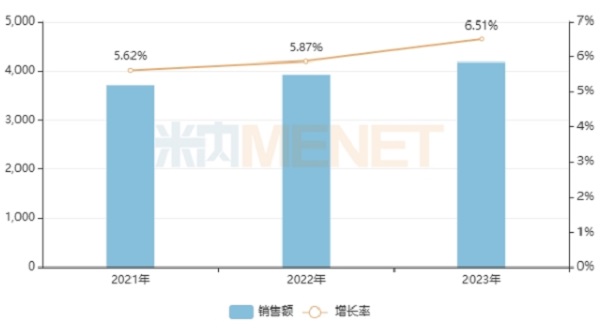
图1:近三年中国三大终端六大市场中成药的销售规模(亿元)
来源:米内网格局数据库
中国中医科学院中药研究所资深研究员、北京盈科瑞创新医药股份有限公司董事长张保献提到,目前中医药事业在中医药传承与创新政策推动下不断发展,成绩已有目共睹,但人民群众对中医药的要求正在不断提高,国家推进培育以更显著临床价值为导向的新质生产力,这些内因外因促使着中医药走向更快提升并真正实现中西医并重的新局面。
中药的临床价值已逐渐成为中医药高质量发展的核心,中国医学科学院药用植物研究所原所长孙晓波认为“从临床中来,到临床中去,建立中药上市后评价关键技术体系很重要”,这也是临床价值进一步挖掘的体现。中成药大品种具有多成分、多途径和多靶点的作用特点,基于药物的核心功效,通过网络药理学、整合药理学和生物信息学等技术建立中成药大品种药效机制评价体系,进而利用这一体系评价中成药大品种对重大疾病的治疗作用,并深入研究其作用机制,找出药效关键靶点与信号通路,突出中成药大品种的整体效应,体现中成药大品种的临床价值。
中华中医药学会发布的《新时代中医药标志性科技成果(2012-2022)》中提到,中药二次开发与智能制造推动着中药产业高质量发展,其成果促进了中药产业技术革新,也推动了中药产业智能化技术升级。完善中药全产业链技术,挖掘中药潜在价值,助力二次开发,促进中药大品种不断涌现,助力中药市场不断扩大。
“古方新药”大有可为,立足国内走向国际
我国中药有着几千年历史,人类使用经验丰富,青蒿素的发现也是得益于中医药使用青蒿抗疟疾的历史,古方引领着新药研发思路,已成为近几年中药研发领域的最热话题。
2020年9月发布的《中药注册分类及申报资料要求》提到,古代经典名方是指至今仍广泛应用、疗效确切、具有明显特色与优势的古代中医典籍所记载的方剂,而古代经典名方中药复方制剂是指来源于古代经典名方的中药复方制剂。据《中华人民共和国中医药法》第三十条提到,生产符合国家规定条件的来源于古代经典名方的中药复方制剂,在申请药品批准文号时,可以仅提供非临床安全性研究资料。
北京盈科瑞创新医药股份有限公司科研总裁李艳英提到,按照国家药监局发布的《中药注册管理专门规定》,古代经典名方复方制剂可豁免药效学和临床试验研究申报上市,较大程度降低了研发费用,一般可控制在400-600万。由盈科瑞全面负责技术研究和注册申报的3.1类古代经典名方中药复方制剂枇杷清肺颗粒(敖东洮南药业)于 2023 年1月提交上市中请并获得受理,审评过程顺利,零发补,圆满地通过了技术审评和现场核查,仅仅6个月时间便顺利批准上市(2023年7月),此外,一贯煎颗粒(神威药业集团)从报产到获批(2023年7月-12月)也是经历了5个月左右。目前盈科瑞还有二冬汤颗粒、升陷颗粒等7个古代经典名方中药复方制剂报产在审,有望在今年年底及明年陆续获批,成为市场的新宠儿。
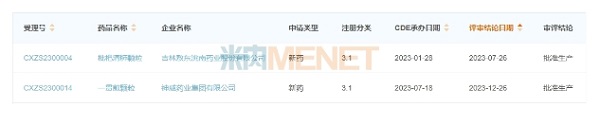
图2:盈科瑞研发的枇杷清肺颗粒和一贯煎颗粒申报情况
来源:米内网中国申报进度(MED)数据库
香港浸会大学药学博士&华药君康高级顾问黄韬认为,中药复方是体现中医药学传统理念和智慧的典型代表,也是目前中医药走向国际化和规范化的关键环节。他提到在研项目CDD-2101,是以麻子仁丸为基础进行技术改良,强化从药材到中间体到成品的质量控制,适用于治疗便秘(包括但不限于肿瘤患者、肥胖人群及帕金逊症病人的便秘等),已符合美国FDA对植物药研发的要求,是第一款在香港研发并获U.S. FDA批准进行临床试验的中药。他表示,通过循证医学的方法展示传统中医药临床疗效、扩展临床用途,为古方新药的现代研发提供基础,一直是中医药研究者面临的挑战。
提及美国FDA对植物药研发的要求,香港浸会大学中医药研发中心科学顾问窦金辉根据“植物药开发指南”精神和已经能上市植物药NDA/BLA,提出了自己的见解。他认为中药(包括复方)成为新植物药NME(新分子实体)上市,需要积累大量CMC切实做到质量可控,包括批次间质量参数的相对稳定(即有可被监管部门接受的区间范围);早期临床的非临床(药理和毒理)要求,虽然可用中药人用经验给予支持得到相对灵活的处理,但是GLP毒理对后期临床试验(如III期)和新药注册申请是必须的;研发还要排除或者预见可能的安全隐患(包括大剂量的毒性靶器官等)并关注特殊受试人群临床用药安全(如肝肾功能不全,婴幼儿,孕妇等);新药上市需要有对照的和设计良好的临床试验支持安全有效和使用标签的要求对植物药和其药品是一样的。他希望在推动中药植物药的发展时,选择合适的候选药物,并在CMC、药理、毒理以及临床试验中,把中药以处方药的形式进行研究,然后进入美国并继而走向世界。
新市场新商机!粤港澳大湾区潜力可期
2019年2月《粤港澳大湾区发展规划纲要》提出建设宜居、宜业、宜游的优质生活圈,塑造健康湾区,粤港澳大湾区已逐步成为中国开放程度最高、经济活力最强的区域之一,在国家发展大局中具有重要战略地位。
近几年,粤港澳三地在药械发展上不断有新政出台:2020年9月《粤港澳大湾区药品医疗器械监管创新发展工作方案》,2021年7月澳门特区政府发布《中药药事活动及中成药注册法》,2021年8月《广东省粤港澳大湾区内地临床急需进口港澳药品医疗器械管理暂行规定及其配套制度规范》及《广东省药品监督管理局关于简化在港澳已上市传统外用中成药注册审批的公告》,2022年6月《支持港澳医疗器械注册人在大湾区内地9市生产医疗器械实施方案》及《支持港澳药品注册人在大湾区内地9市生产医疗器械实施方案》,2023年8月《关于支持在横琴粤澳深度合作区使用澳门地区已上市部分药品的工作方案》,2023年10月成立“香港药物及医疗器械监督管理中心”……系列政策不仅进一步提升粤港澳大湾区在国家经济发展和对外开放中的支撑引领作用,支持香港、澳门融入国家发展大局,也给予了中药进入港澳地区乃至出海提供了便利。
粤港澳大湾区药品与医疗器械真实世界研究院院长蒋杰分析了澳门中药注册政策的亮点:首先,实行中成药注册持有人制度(MAH),对于澳门或横琴研发的中成药,允许自然人及法人申请中成药;其次,可对照在任何国家或地区注册或取得销售许可的中成药开发同名同方药;第三,对照《中国药典》品种开发的同名同方药,无需对照药对比研究数据、药理毒理数据和临床研究数据;第四,创新药已有人用经验证据能预测临床价值、可满足中医临床需求但使用范围不涉及危重症,临床拟用剂量已有人用经验的,且毒理学研究未发现明显毒性,可仅提供一般文件、药学研究资料、毒理学及III期临床资料;第五,创新药为已有5年或以上的使用经验或具有300例以上完整临床病历的医院制剂,且中成药的配方、工艺、剂型及临床应用等与该医院制剂一致,可仅提供一般文件、药学研究资料、毒理学及III期临床资料(300例);第六,创新药来源于中国工程院、中国科学院院士(临床专业)或国医大师,且能提供临床试验剂量探索、临床定位、适用人群、疗程探索的研究资料,同时拟用剂量获得药物重复给药毒性研究结果支持的,可仅提供一般文件、药学研究资料及III期临床研究资料;第七,审批时间更短。
此外,结合港澳外用中成药上市多年、群众惯用广泛和外用剂型安全性相对较高的特点,港澳外用中成药注册审批流程得到了简化,缩短外用中成药进口上市审批时间,满足粤港澳大湾区居民用药需求的同时,积极支持港澳中药产业发展,推动粤港澳大湾区中医药融合发展。香港、澳门本地登记的企业持有,并在港澳地区经批准上市且使用五年以上的传统外用中成药,可以简化申请进口粤港澳大湾区。通过粤港澳药品监管机制对接,减少注册审批过程中的生产现场检查和体系核查环节。规定港澳外用中成药上市后变更应当参照内地有关药品上市后变更管理要求及技术指导原则进行研究验证。如在港澳获得批准的,可申请调整审批流程改为备案流程办理。上市注册审批的技术审评由原来的200个工作日缩减至80个工作日办结,审批总时限由原来的235日减至115日;上市后变更审批和再注册时限也分别减少50日,加快审批进程。港澳外用中成药注册申报可提供原在港澳上市注册时提交的试验研究数据作为相应申报数据(但不代表不需要补充其他研究数据)。以港澳地区上市使用实际情况作为重要证据,可以不提供药物临床试验资料,改由广东省药品监管局组织对产品在港澳地区上市使用情况及不良反应收集情况进行综合评估。至今已有13个港澳外用中成药获得进口注册许可,同时有十几个品种在申报或准备申报。
中药进入港澳地区更加便利,同时也为医药国际化带来了新机遇,蒋杰提到“引进来”和“走出去”两大方面,供中药企业做战略参考。引进港澳地区知名的外用药品牌,不仅丰富了产品线,还能实现全国销售。提前引进香港口服中成药,静候大湾区放开港澳口服中成药简化进口注册政策,有望迎来新商机。创新药械进口注册和大湾区医院引进“两条腿”一起走路,提前进入国内市场,积累真实数据和使用案例。至于走出去,巧用澳门及香港中药注册制度的差异点,以及利用港澳窗口开拓产品一带一路及国际市场,加快产品成果转化。粤港澳大湾区不仅是国家的重要战略布局,也将成为中药发展的新版图。
注:米内网《中国三大终端六大市场药品竞争格局》,统计范围是:城市公立医院和县级公立医院、城市社区中心和乡镇卫生院、城市实体药店和网上药店,不含民营医院、私人诊所、村卫生室,不含县乡村药店;上述销售额以产品在终端的平均零售价计算。
